產品和服務

分子模擬代算
高性能分子模擬工作站,帶您走進理論計算的大門

量子化學代算
高性能原子模擬工作站,帶您走進理論計算的大門

分子對接服務
提供各種軟件的分子對接業務,剛性,柔性,半柔性對接

分子建模服務
各種分子定制建模服務,包括粗粒化模型,原子模型

機器學習與虛擬篩選
QSAR,基于機器學習的預測篩選





高性能分子模擬工作站,帶您走進理論計算的大門
高性能原子模擬工作站,帶您走進理論計算的大門
提供各種軟件的分子對接業務,剛性,柔性,半柔性對接
各種分子定制建模服務,包括粗粒化模型,原子模型
QSAR,基于機器學習的預測篩選

分子動力學模擬
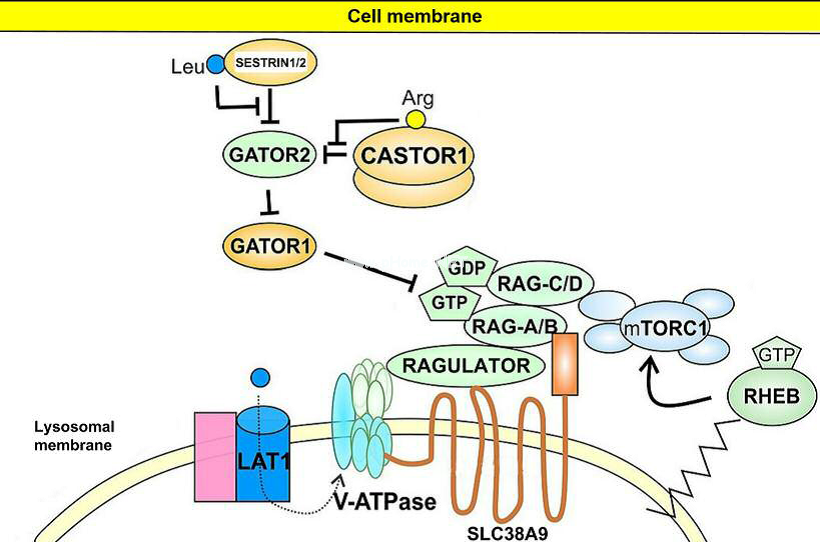
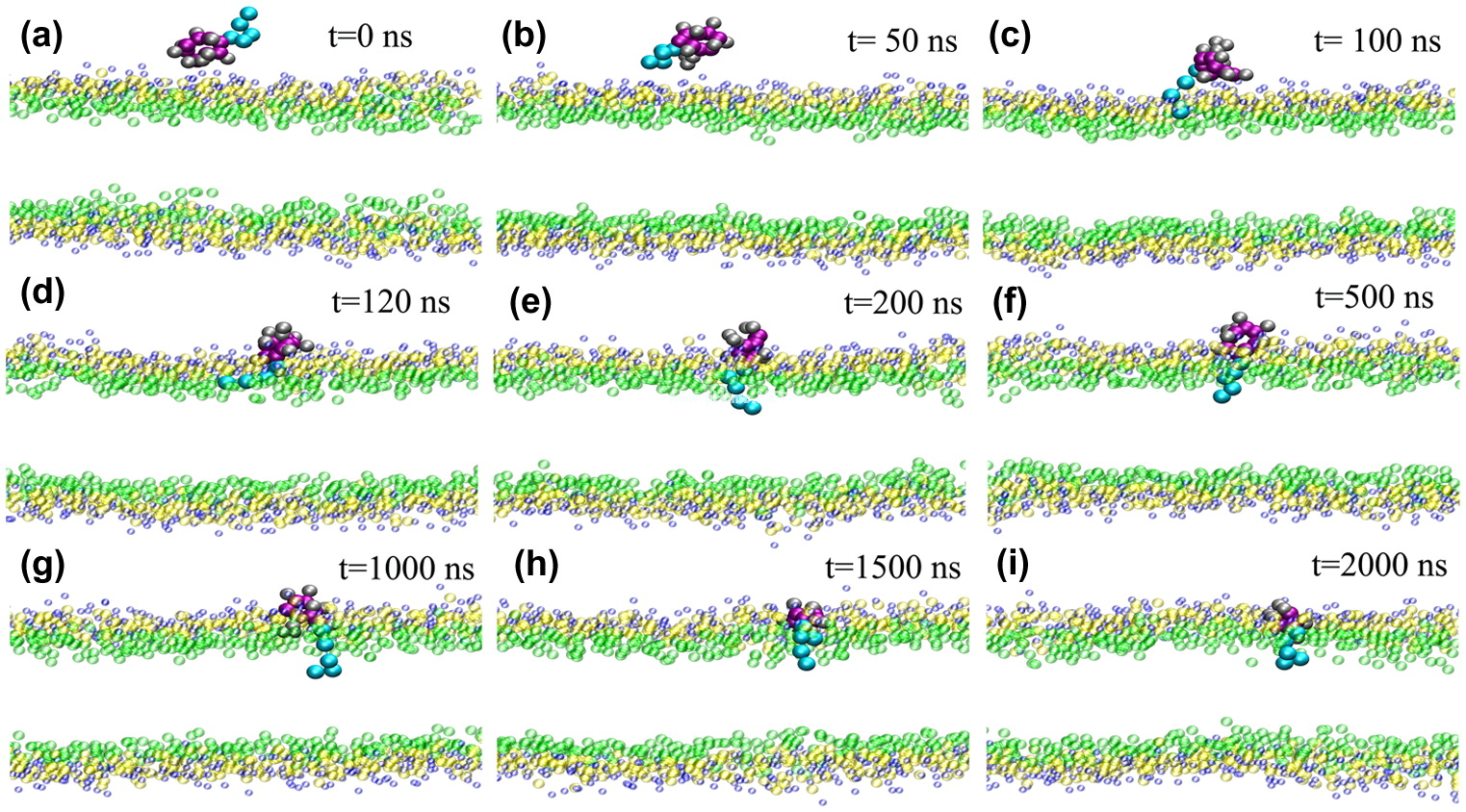
| 分子動力學(Molecular Dynamics,MD)模擬即通過傳統牛頓力學,在考慮分子間相互作用力和分子內的原子間的成鍵作用,以及與溶劑之間的親疏水作用下,通過迭代更新每個粒子的新的位置和速度從而對反應進行模擬,通常外部設置NPT或NVT系宗來模擬實驗條件,在進行能量最小化之后使得整個體系達到平衡,對平衡后再進行模擬一定時間尺度,最終通過對平衡后的軌跡進行統計分析每個原子或者整體結構的變化,最終對某些現象給出分子、原子尺度的解釋。
分子動力學模擬主要包括以下具體內容: |
|
| 所提供的分子動力學模擬類型
· 研究各種蛋白、RNA、DNA與各種有機、無機化合物的復合物體系的相互作用機制: · 蛋白受體-小分子化合物配體之間的作用機制; · 細胞膜、脂類化合物與多肽、天然化合物的相互作用在分子層面的機理; 其他高級方法: · Slab單板動力學模擬; · 定向拉伸動力學模擬; · 金屬表面材料模擬; · 石墨烯等二維材料有關模擬; · 樹狀、球型等非常規形狀模擬。 |
針對模擬結果進行分析:
分子構象變化:RMSD、RMSF; 量子化學QM/分子動力學MM結合: |
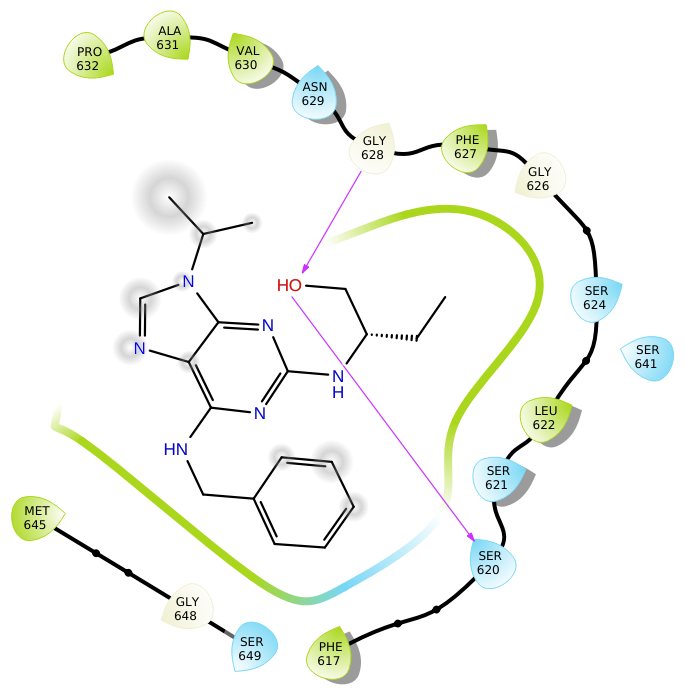
分子對接
| 分子對接(Molecular Docking)通常是指將兩個分子(比如蛋白-蛋白分子,蛋白-小分子化合物)通過算法改變其中一個分子的局部結構或者分子骨架并計算其復合物的能量大小,并對能量大小進行排序,從而實現特定分子結 構的優化,得到更具有活性的結構,通過對數據庫中大量分子進行對接,進而實現虛擬篩選,常用于靶向藥物篩選、設計,材料化合物結構定向優化等。
不同的分子對接軟件側重不同體系的對接: |
|
| 蛋白質與小分子體系
蛋白質-小分子化合物體系常使用AutoDock vina進行對接。
|
其他體系對接
包括蛋白與蛋白(優化側鏈)、抗體-抗原、蛋白質-多肽、蛋白質-表面對接、離子-離子對接等體系
|
定量構效關系
定量構效關系(QSAR)即利用結構的信息來預測候選分子活性的模型,是基于配體的另一種虛擬篩選方法,通過研究基于生物活性變化與一組化合物中的結構和分子變化相關聯,從相關性產生統計模型,以開發數學模型預測新型化合物的生物學特性。類似的方法在材料結構-性質方面的預測稱為QSPR模型。QSAR廣泛應用于藥物研發、催化反應等活性分子的合理設計中。
QSAR主要有以下方法:
| 3D-QSAR及以下傳統方法 | 4D-QSAR及以上與機器學習方法 |
| 一維度描述符:原子計數、鍵計數、分子量、 二維描述符:MACCS、MOL2、拓撲指數、分子輪廓和二維自相關描述符、化學指紋 三維描述符:CoMFA、CoMSIA即比較分子場方法和比較分子相似性方法 |
4D:考慮多種結構構象的三維化學描述符的擴展 5D:考慮諸如受體柔性和誘導契合的因素 6D:考慮到溶劑化作用在受體-配體主要相互作用中的影響 機器學習方法:使用大量數據,通過手動或者自動特征提取對訓練集進行模型訓練,從而使用模型進行新的化合物活性預測。 對候選化合物可提取的特征包括顯式特征:對分子概況,取代基團/官能團,物理化學性質,非鍵相互作用特征,特殊功能片段,帶電性,親疏水基團,氫鍵,立體幾何描述符,拓撲特征,動態配體相互作用特征,量化特征,其他描述符;隱式特征:通過神經網絡、矩陣分解獲得的表征 |
3D-QSAR
3D-QSAR(Pharmacophore)是從化合物三維坐標表示中提取化學特征,被認為是對結構變化最敏感的。3d指紋是基于藥理學模式、表面性質、分子體積或分子相互作用場的化學特征。
各種最新的方法:
最著名的三維指紋之一是分子相互作用場(MIF),基于MIF的指紋可以通過推導三維網格點和復合物活性之間的關系,被用于CoMFA
應用最廣泛的3D-QSAR方法是CoMFA和CoMSIA即比較分子場方法和比較分子相似性方法
Schrodinger軟件包提供PHASE作為其3D-QSAR功能。
使用互補生物目標場來改進基于配體的3D QSAR模型的兩種方法是COMBINE和AFMoC計劃
另外兩種開創性的3D QSAR方法,HASSLE和MSTD也值得一提
最新的3D-QSAR策略包括Topomer CoMFA、比較結合能分析法(Comparative Binding Energy,COMBINE)、比較分子表面分析法(Comparative Molecular Surface Analysis,CoMSA)和比較殘基相互作用分析法(CoRIA)
2D/3D相似性
分子結構相似性搜索是基于配體發現藥物的常用技術。其目的是識別并返回結構和生物活性與查詢化合物相似的候選化合物。兩個分子的結構相似性通常通過計算它們的tanimoto系數(Tc)來評估。化學指紋Tc,也被稱為jaccard索引,是對集合之間相似性的度量,這些集合將相似性分數計算為兩個特征向量共享的位的分數。高Tc值表明兩種化合物相似,但不提供相似的信息維度,例如它們共享的特定化學基團。
有關方法:
3D Tc是一種常見的3d相似性度量,用于計算兩個比較配體之間共享分子體積的分數。
基于體積的相似性實現的例子包括化學結構快速重疊(ROCS)程序
基于分子形狀高斯表示的藥物發現中最流行的形狀相似性方法
另一個3D相似性度量是藥理學相似性,它只考慮關鍵功能組之間的體積重疊,與藥理學相似性相關的一個類似概念是在FieldAlign工具
Lo等人開發了shapeAlign程序,該程序結合了基于Obabel PF2指紋、形狀和藥物學點的二維和三維指標,用于無監督的三維化學相似性聚類
費雷拉等開發了一種新的相似性度量,稱為化學語義相似性,以根據化學化合物的語義特征(如ChEMBL數據庫中的藥物注釋)對其進行分類
匹配分子對(MMP)形式主義已經出現,作為一種定義特定類型的轉換或關系、非環單鍵取代和促進索引和搜索模擬關系方法的發展
Hussain和Rea開發的分段索引算法是目前最廣泛使用的MMP搜索方法,但不支持相似性搜索。
Rensi和Altman開發了一種使用Tanimoto內核嵌入指紋計算化學轉化相似性的方法,并將模糊搜索功能擴展到MMP框架
同源模建
同源模建(Homology Modeling)即通過使用多個蛋白質化合物的已知三維晶體結構來為一個僅知道蛋白序列而未知其三維結構的蛋白產生可能的三維結構,用于已知蛋白二維序列,但尚未得到其三維晶體結構的場景。
| 建模處理過程例子 |
| l 使用Clustal Omega通過多重序列對比找到合適的已知對應晶體結構的多個蛋白; l 使用RosettaCM進行多模板建模; l 對產生的晶體結構進行動力學優化; l 通過分子對接或者動力學模擬進行評估。 |
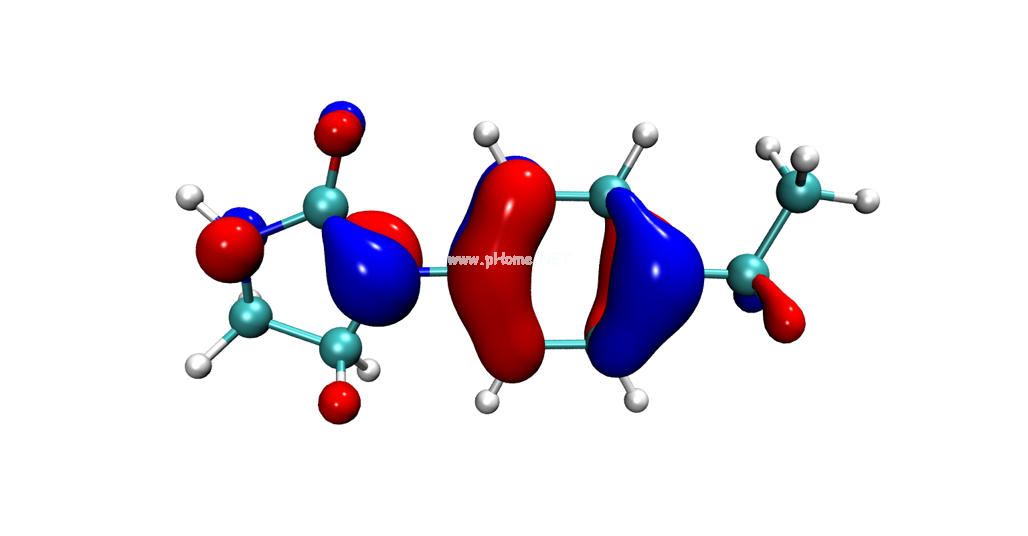
量化計算
量子化學(Quantum Chemistry)計算是使用量子化學方法比如DFT、波函數方法來模擬原子之間的化學反應機理。通過引入量子化學計算,可以使用電子結構模型來研究一系列的化學現象,探究其化學本質。
采用量化計算技術,主要進行以下方面的研究:化學反應機理、激發態反應、分子性質預測、弱相互作用、酶催化機理、反應過渡態搜索、反應產物預測、化學性質預測、無機催化機理、分子軌道、波譜預測(IR、UV-Vis、NMR、熒光光譜、ECD、VCD、ROA、ORD、EPR/ESR)。
虛擬篩選
虛擬篩選(Virtual Screening)即利用計算機技術輔助藥物或者材料開發。
虛擬篩選涉及技術包括同源建模、分子對接、定量結構-活性關系模型和分子動力學模擬等傳統篩選技術。當前以大數據和高性能計算為基礎的機器學習技術在大幅提高藥物開發效率方面已經表現出巨大潛力,尤其是藥物發現方面。
專門為制藥公司提供服務的機器學習公司激增,本公司也是其中之一。主要提供包括疾病目標靶點識別、化合物篩選、從頭開始的藥物設計以及臨床圖像識別、毒性和ADME的預測等上游技術,又稱為CRO技術公司。
我們在虛擬篩選方面有三大優勢:
 483
550
wolf
wolf1583307687 1583307687
483
550
wolf
wolf1583307687 1583307687
 483
550
wolf
wolf1583307681 1583307681
483
550
wolf
wolf1583307681 1583307681
 483
550
wolf
wolf1583307420 1583307420
483
550
wolf
wolf1583307420 1583307420
 483
550
wolf
wolf1583307199 1583307199
483
550
wolf
wolf1583307199 1583307199
 483
550
wolf
wolf1583272322 1583272322
483
550
wolf
wolf1583272322 1583272322
 483
550
wolf
wolf1583269120 1583269120
483
550
wolf
wolf1583269120 1583269120

基于Martini力場制作樹狀分子的粗粒化力場

模擬高分子在水溶液的自組裝行為

到校實體培訓三天分子動力學在纖維素,蛋白質-小分子中的應用

租用我公司服務器利用Gromacs程序計算有機分子的擴散系數

計算離子液體在晶體表面的擴散系數,吸附能

計算抗凍蛋白與冰晶相互作用的機理研究
我們具有創新的思維,已完成200多個富有挑戰性的項目方案。
我們有專業的技術和豐富的經驗,多名工程師已在本行業工作10年。
我們高效地工作,一年365天在線,加急的項目可在3天內完成項目報告
我們以專業的態度做適用的解決方案,致力于成為分子模擬技術行業優秀的服務商。 術理科技擁有一套完整的從分子模擬研究到化學合成、活性測試的技術服務體系
點擊給我發消息

掃碼添加微信
Copyright ? 2021術理科技有限公司 版權所有 粵ICP備19082003號